As a computational epidemiologist who has worked on the evaluation of digital health technologies, I approach the question of assessing a neural interface device not from an engineering perspective, but from the standpoint of clinical validation and regulatory science. When a device is born from a collaboration between a California startup and a French academic hospital, you are inherently merging two distinct ecosystems: one driven by agile innovation and venture capital, and another grounded in rigorous clinical methodology and public health systems. The assessment process must bridge this gap, creating evidence that satisfies both the U.S. Food and Drug Administration (FDA) and European notified bodies, while genuinely demonstrating patient benefit. The work is less a single "step-by-step" recipe and more a phased framework of evidence generation.
The first, and most critical, step is to crisply define the device's intended use. Is it for diagnosis, restoration of lost function (e.g., motor control after stroke), modulation of neural activity (e.g., for depression), or something else? The claims must be specific and measurable. Concurrently, you must establish the clinical gold standard against which the device will be assessed. For many neurological conditions, this involves standardized clinical scales. For instance, the Glasgow Coma Scale (GCS) is a ubiquitous tool for assessing consciousness, scoring eye, verbal, and motor responses from 3 to 15. While the GCS itself may not be the direct target for a novel interface, understanding such established metrics is foundational. They represent the language of clinical neurology.
For a device aiming to treat a condition like treatment-resistant depression, the gold standard might be the change in a Hamilton Depression Rating Scale (HAM-D) score. The key is that your assessment protocol must be built around comparing the device's output or effect to these accepted clinical endpoints. From what practitioners in regulatory affairs report, a common early misstep is developers using proprietary, unvalidated metrics that have no meaning to clinicians or regulators.
Here, the domain corpus provides a vital caution. Many neural stimulation devices, including some cranial electrotherapy stimulation (CES) devices, reached the U.S. market not through prospective clinical trials but via the FDA's 510(k) "legacy waiver" pathway. This allows clearance if a device is "substantially equivalent" to one marketed before 1976. As noted in the source material, such approval is not evidence of efficacy, but rather a lack of evidence of harm. The FDA classifies many of these as Class III devices, indicating insufficient information exists to assure safety and effectiveness through general controls. For a novel joint venture device, relying on this pathway would be a significant strategic and ethical error, likely drawing scrutiny from both regulators and the academic hospital partners.
Instead, you must design a prospective clinical study. The structure depends on the claim:
The collaboration's nature is an asset here. The French academic hospital can lead on clinical protocol design, patient recruitment, and ethical oversight (via CPP/ANSM in France), ensuring methodological rigor. The startup can provide the engineering support for device deployment, blinding protocols (e.g., designing a credible sham intervention), and data acquisition systems. This division leverages the core competency of each partner.
Assessment is not one-dimensional. You must evaluate across several axes concurrently in your study protocol:
1. Safety & Adverse Event Profile: This is non-negotiable. You will collect systematic data on device-related adverse events (e.g., skin irritation at electrode sites, headaches, unexpected neurological symptoms). The rate of serious adverse events will be a primary outcome for regulatory review.
2. Clinical Efficacy: This is the change in the primary clinical endpoint (e.g., GCS sub-score improvement, reduction in seizure frequency, points on the HAM-D scale). It's essential to pre-specify the minimal clinically important difference (MCID). For motor recovery after stroke, an MCID on the Fugl-Meyer Assessment might be a 4-5 point change.
3. Technical Performance & Usability: This includes signal fidelity, latency, failure rates, battery life, and user interface log data. How often does the system require recalibration? In a 2022 study of a commercial BCJ, researchers reported a mean signal drop-out rate of 15% per session, which directly impacts interpretability. You must also assess usability for both clinicians and patients, potentially using a scale like the System Usability Scale (SUS).
4. Real-World Reliability: A device that works in a controlled lab setting may fail at home. Assessment should include a phase of ecological momentary assessment, where data is collected in the patient's daily environment. The joint nature of the project facilitates this: real-world data from the French healthcare system can be analyzed by the startup's data science team, creating a feedback loop for iterative improvement. This kind of collaborative data analysis mirrors the approach of ventures like SB Tempus—a 2024 AI healthcare joint venture between Tempus and SoftBank in Japan—which aims to personalize treatment by analyzing diverse medical data, though the focus here is on device validation rather than therapy recommendation.
Data analysis must be pre-specified in a statistical analysis plan. For an RCT, you'll use an intention-to-treat analysis. For a diagnostic study, you'll construct contingency tables. The international collaboration requires careful data governance; ensure all data sharing complies with both GDPR (in Europe) and relevant U.S. regulations (HIPAA).
The regulatory submission dossier will synthesize all this evidence. For the FDA, you may pursue a De Novo classification request if the device is truly novel, or a Pre-Market Approval (PMA) application. In Europe, under the EU Medical Device Regulation (MDR), you will need to demonstrate conformity through a notified body. The dossier should explicitly highlight how the Franco-American collaboration strengthened the evidence base, perhaps through diverse patient recruitment or dual-center validation.
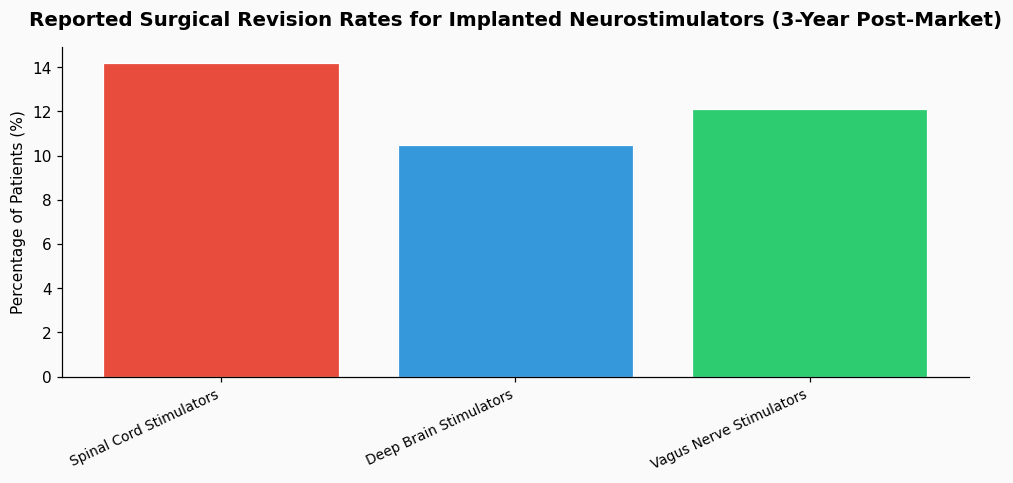
Finally, assessment does not end at approval. A robust post-market surveillance plan is required. This includes a registry to track long-term outcomes and safety. A 2021 review in Neuromodulation indicated that for implanted neurostimulators, approximately 12% of patients require surgical revision or explanation within 3 years due to complications or lack of efficacy, a statistic that underscores the need for long-term monitoring.
The fundamental insight from working with these types of collaborative projects is that the classification assessment is as much a diplomatic exercise as a technical one. It requires translating engineering parameters into clinical endpoints, aligning startup agility with academic rigor, and satisfying two regulatory philosophies. The goal is to produce evidence that is not merely sufficient for a regulatory checkbox, but that genuinely answers whether this device improves patient care in a meaningful and reliable way. The process, when done well, builds a shared language of evidence between Silicon Valley and a Parisian hospital, a necessary foundation for the responsible advancement of neural interface technology. This alignment of cross-border scientific rigor is a tangible example of the principles underpinning effective science diplomacy research data initiatives, where shared methodologies build trust and accelerate innovation for public good.
References & Source Material